
Learning Objectives
- Compare and contrast genome components, size and organization of prokaryotic versus eukaryotic genomes
- Explain why genome size corresponds to metabolic complexity in prokaryotes but does not predict organismal complexity or phylogeny in eukaryotes.
- Contrast the proportion of protein coding DNA and other types of DNA in a representative mammalian genome
- Describe the current and potential applications of genome sequencing technology
One of the defining and essential features of life is genetic material. An organism’s genome is the complete set of all genes and genetic material that is present in that organism or individual cell. Often we think of genes in terms of protein-coding genes, or genes that are transcribed into mRNAs and then translated into protein; however, genomes consist of a lot more than just protein coding genes. In addition, the features of prokaryotic and eukaryotic genomes differ in terms of both size and content.
The image below shows the different ranges of genome sizes in different taxonomic groups of life. Note that, in general, prokaryotic genomes are smaller than eukaryotic genomes. However, eukaryotic genome sizes vary wildly and are not linked to organismal “complexity.” Refer to this diagram as you read on about the differences and similarities between prokaryotic and eukaryotic genomes.

Prokaryotic Genomes
- The genomes of Bacteria and Archaea are compact; essentially all of their DNA is “functional” (contains genes or gene regulatory elements).
- The sizes of prokaryotic genomes ranges from about 1 million to 10 million base pairs of DNA, usually in a single, circular chromosome
- Genes in a biochemical pathway or signaling pathway are often clustered together and arranged into operons, where they are transcribed as a single mRNA that is translated to make all the proteins in the operon.
- The size of prokaryotic genomes is directly related to their metabolic capabilities – the more genes, the more proteins and enzymes they make.
Eukaryotic Genomes
- The genome sizes of eukaryotes are tremendously variable, even within a taxonomic group (so-called C-value paradox).
- Eukaryotic genomes are divided into multiple linear chromosomes; each chromosome contains a single linear duplex DNA molecule.
- Eukaryotic genes in a biochemical or signaling pathway are not organized into operons; one mRNA makes one protein.
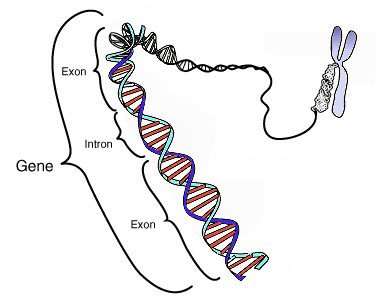
- Many eukaryotic genes (most human genes) are split; non-coding introns must be removed and the exons spliced together to make a mature mRNA. Introns are “intervening” sequences in genes that do not code for proteins. The image below shows a zoomed-in region of a gene highlighting the alternating exons and introns.
- The multiple exons in a eukaryotic gene can be spliced in different ways to make multiple mRNAs and multiple proteins from a single gene (alternative splicing).
- The majority of human genes can be spliced in two or more different ways. Therefore, the actual number of human proteins far exceeds the number of protein-coding genes.
- Alternative splicing often results in “tissue-specific” versions of the same gene, where one splice variant is present in, for example, cardiac muscle, while a different splice variant of the same gene is present in skeletal muscle. The image below shows one hypothetical gene with 3 different possible proteins depending on which exons are included in the final mRNA.

What accounts for the variation in genome size?
There is no good correlation between the body size or complexity of an organism and the size of its genome. Eukaryotic genomes sequenced thus far have between ~6,000 and ~30,000 protein-coding genes, or less than 10-fold variation in the number of genes. The human genome has just over 19,000 protein-coding genes (recently revised down from 21,000 genes). Therefore, the 10,000-fold variation in eukaryotic genome size is due mostly to varying amounts of non-coding DNA.
Here is a quick comparison of the genome size and predicted gene number for a sampling of eukaryotes:
| organism | chromosome number (diploid) | base pairs | predicted number of genes |
| Saccharomyces cerevisiae (budding yeast) | 16 | 1.25×107 | 6,275 (~5,800 functional) |
| Drosophila melanogaster (fruit fly) | 8 | 1.65×108 | 13,600 |
| Caenorhabditis elegans (nematode worm) | 6 | 1.0×108 | ~19,000 |
| Canis familiaris (dog) | 78 | 2.4×109 | ~19,000 |
| Homo sapiens (human) | 46 | 3.3×109 | ~19,000 |
| Mus musculus (mouse) | 40 | 3.4×109 | ~20,000 |
| Oryza sativa (rice) | 24 | 4.66×108 | ~37,000 |
It’s very interesting to note that humans have about the same number of genes as the microscopic nematode worm, C. elegans, and fewer genes than rice.
What’s in the human genome?

- Protein-coding (exon) DNA sequences comprise less than 2% of the human genome.
- Introns make up just over 1/4 of the human genome.
- Transposable elements and DNA derived from them make up about 1/2 of the human genome. Transposable elements are essentially “parasitic” DNA that resides in a host genome, taking up space in the genome but not contributing useful or functional sequences to the genome. They are the DNA transposons, LTR retrotransposons, LINEs and SINEs.
- Because they are parasitic DNA elements, transposable elements are extremely valuable for studying evolutionary relationships. If a transposable element “invades” an organism’s genome, then it is likely to remain in that genome as the population evolves and when speciation occurs. If the same transposable element is present in the same location in the genomes of two different species, this is strong evidence that those two species share a recent common ancestor who also had the transposable element in its genome.
- One family of SINEs, called the Alu element, is a 300-nucleotide sequence that is present in over 1 million copies in human and chimpanzee genomes.
- Segmental duplications are relatively long (> 1 kb; kb = 1,000 bp) segments of DNA that have become duplicated. These duplications create copies of genes that can mutate and acquire new functions. Gene families (e.g., alpha- and beta-hemoglobin, myoglobin) arose this way.
Is the human genome 80% “junk” or 80% functional?
Recent publication of data and papers from the ENCODE project, a systematic survey of the human genome variation and activity from chromatin modifications to transcription, has claimed that, contrary to previous belief, fully 80% of the human genome has at least some biochemical activity, such as transcription (The ENCODE Project Consortium, 2012). Indeed, many small RNAs, called microRNAs (miRNAs) with important regulatory roles are transcribed from intergenic regions. However, these miRNAs and other regulatory RNAs comprise less than 1% of the human genome, and other studies have indicated that only 10% of the genome appears to be subject to some evolutionary constraint (review by Palazzo and Gregory, 2014).
DNA sequencing

The human genome project was accomplished by large banks of automated sequencers that used the Sanger dideoxy sequencing technology. In recent years, however, massively parallel sequencing technologies have brought down the cost and throughput of DNA sequencing much faster than computing speed and power has increased (Moore’s Law).
The implications for being able to obtain huge amounts of DNA sequence quickly and cheaply has startling implications for biological research in all fields, and for human health. The TedTalk below by Richard Resnick discusses some of the applications:

Put it all together
DNA sequencing and personal genomics case study
B1510_module4-7_Genomes__questions_2012
How genomics is affecting people
The story of two Georgia Tech grads, and how DNA sequencing and social media launched discovery and research of a previously unknown genetic disease: http://www.newyorker.com/magazine/2014/07/21/one-of-a-kind-2
A story in Atlantic (9/2015): How Genome Sequencing Creates Communities around Rare Disorders
Additional resources
Ralston, A. (2008) Operons and prokaryotic gene regulation. Nature Education 1(1)
Sustainable Development Goal

UN Sustainable Development Goal (SDG) 17: Partnerships for the Goals – Collaboration between researchers, industry, and policymakers is essential for ensuring the responsible use and application of genome information and technologies. Further, such partnerships help to ensure the long-term viability of the programs by increasing the capacity of industries and people through education and economic investment.

http://www.ted.com/talks/jennifer_doudna_we_can_now_edit_our_dna_but_let_s_do_it_wisely
In the wikipedia article on operons, it says that eukaryotic genomes have recently (1990s) been discovered to have numerous instances of polycistronic transcription
—> http://bfg.oxfordjournals.org/content/3/3/199
Polycistronic transcripts in eukaryotes are exceptional – “numerous” is misleading. For the purpose of an intro biology course, as a general rule eukaryotes do not have polycistronic transcripts. Eukaryotes have tremendous diversity, and some eukaryotic organisms do have a few polycistronic transcripts, which are still exceptional even for those organisms.
Nice article on interaction of genes with drug metabolism
http://well.blogs.nytimes.com/2016/07/12/for-coffee-drinkers-the-buzz-may-be-in-your-genes/